London boy receives heart transplant after ultra-rare LMNA diagnosis
Great Ormond Street case highlights N-of-few medicine, donor organs and research funding stay scarce
Images
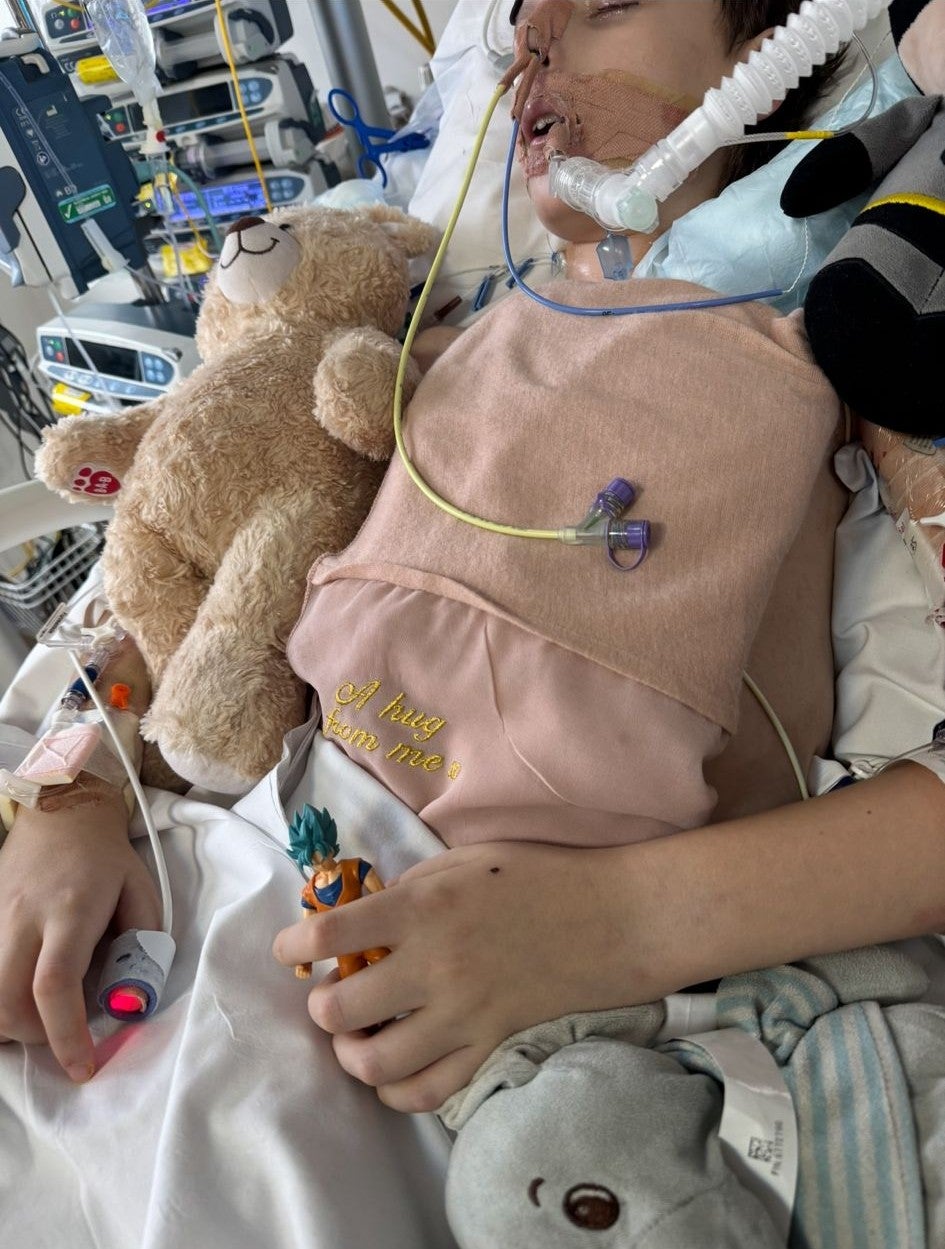 Trey was in a coma for six weeks (Supplied)
Supplied
Trey was in a coma for six weeks (Supplied)
Supplied
 Trey celebrated his one year anniversary of getting a heart transplant (Supplied)
Supplied
Trey celebrated his one year anniversary of getting a heart transplant (Supplied)
Supplied
 Trey’s mother has called for more people to become organ donors (Supplied)
Supplied
Trey’s mother has called for more people to become organ donors (Supplied)
Supplied
An 11-year-old boy in London received a heart transplant after doctors linked his sudden heart failure to an ultra-rare genetic condition believed to affect only 13 people worldwide, The Independent reports. Trey Taylor spent six weeks in an induced coma and woke to learn he had a new heart and a diagnosis: a rare LMNA gene-related muscular dystrophy affecting skeletal muscle and the heart. His family initially thought he had a stomach virus after he became ill while shopping.
The case is a reminder of how “rare disease” medicine works in practice: a medical emergency first, an explanation later. When a condition is measured in dozens of known patients globally, the usual evidence ladder—large trials, meta-analyses, guideline cycles—does not exist. Care instead depends on specialised centres, rapid escalation pathways, and the ability to interpret genetic findings in context. In Trey’s case, the crucial step was recognising that an enlarged, failing heart in a child could be part of a systemic muscle disorder rather than an isolated cardiac event.
The Independent describes the functional consequences after transplant: Trey uses a wheelchair because the protein in his leg muscles cannot regenerate, but he is not paralysed, and he struggles to explain that distinction to others. That social layer is not incidental. For very rare diagnoses, families often become the long-term translators—explaining the condition to schools, friends and clinicians who may never encounter another case.
The economics around such conditions are structurally awkward. Research budgets and commercial drug development tend to follow patient counts, and the “market size” for a disease affecting a dozen people is effectively zero unless the biology overlaps with more common conditions. Charities and public hospitals therefore become the main funders and organisers of progress: Great Ormond Street Hospital’s charity says it has invested more than £70m into research on rare or complex childhood diseases. Rare Disease Day, which the article uses as a news peg, frames the wider burden differently: an individual disease may be vanishingly uncommon, but rare diseases collectively affect an estimated 300 million people worldwide.
Organ donation is the other scarce input that cannot be scaled by funding alone. Trey’s mother has urged more people to register as donors, describing the transplant as proof that “it saves lives”. For paediatric cases, the constraint is tighter still: matching size and timing can matter as much as surgical skill.
A year after surgery, Trey is alive with a functioning transplanted heart and a genetic diagnosis that may never have a clinical trial. The limiting factor for the next child with the same mutation may be whether the right hospital recognises the pattern quickly enough—and whether a suitable donor heart exists that week.